Spearman-Korrelationskoeffizient#
Der Spearman-Rangkorrelationskoeffizient ist ein nichtparametrisches Maß für die Monotonie der Beziehung zwischen zwei Datensätzen.
Betrachten Sie die folgenden Daten aus [1], die die Beziehung zwischen freiem Prolin (einer Aminosäure) und Gesamt kollagen (einem Protein, das häufig in Bindegewebe vorkommt) in ungesunden menschlichen Lebern untersuchten.
Die unten aufgeführten Arrays x und y erfassen Messungen der beiden Verbindungen. Die Beobachtungen sind gepaart: Jede Messung des freien Prolins wurde aus derselben Leber wie die Messung des Gesamt kollagens am selben Index entnommen.
import numpy as np
# total collagen (mg/g dry weight of liver)
x = np.array([7.1, 7.1, 7.2, 8.3, 9.4, 10.5, 11.4])
# free proline (μ mole/g dry weight of liver)
y = np.array([2.8, 2.9, 2.8, 2.6, 3.5, 4.6, 5.0])
Diese Daten wurden in [2] mit dem Spearman-Korrelationskoeffizienten analysiert, einer Statistik, die für die monotone Korrelation zwischen den Stichproben empfindlich ist und als scipy.stats.spearmanr implementiert ist.
from scipy import stats
res = stats.spearmanr(x, y)
res.statistic
np.float64(0.7000000000000001)
Der Wert dieser Statistik ist tendenziell hoch (nahe 1) für Stichproben mit einer stark positiven ordinalen Korrelation, niedrig (nahe -1) für Stichproben mit einer stark negativen ordinalen Korrelation und gering in der Größe (nahe Null) für Stichproben mit schwacher ordinaler Korrelation.
Der Test wird durchgeführt, indem der beobachtete Wert der Statistik mit der Nullverteilung verglichen wird: der Verteilung von Statistikwerten, die unter der Nullhypothese abgeleitet wurden, dass Gesamt kollagen und freie Prolinmessungen unabhängig sind.
Für diesen Test kann die Statistik so transformiert werden, dass die Nullverteilung für große Stichproben die t-Verteilung von Student mit len(x) - 2 Freiheitsgraden ist.
import matplotlib.pyplot as plt
dof = len(x)-2 # len(x) == len(y)
dist = stats.t(df=dof)
t_vals = np.linspace(-5, 5, 100)
pdf = dist.pdf(t_vals)
fig, ax = plt.subplots(figsize=(8, 5))
def plot(ax): # we'll reuse this
ax.plot(t_vals, pdf)
ax.set_title("Spearman's Rho Test Null Distribution")
ax.set_xlabel("statistic")
ax.set_ylabel("probability density")
plot(ax)
plt.show()
Der Vergleich wird durch den p-Wert quantifiziert: dem Anteil der Werte in der Nullverteilung, die so extrem oder extremer sind als der beobachtete Wert der Statistik. Bei einem zweiseitigen Test, bei dem die Statistik positiv ist, werden sowohl die Elemente der Nullverteilung, die größer als die transformierte Statistik sind, als auch die Elemente der Nullverteilung, die kleiner als das Negative des beobachteten Statistikwerts sind, als "extremer" betrachtet.
fig, ax = plt.subplots(figsize=(8, 5))
plot(ax)
rs = res.statistic # original statistic
transformed = rs * np.sqrt(dof / ((rs+1.0)*(1.0-rs)))
pvalue = dist.cdf(-transformed) + dist.sf(transformed)
annotation = (f'p-value={pvalue:.4f}\n(shaded area)')
props = dict(facecolor='black', width=1, headwidth=5, headlength=8)
_ = ax.annotate(annotation, (2.7, 0.025), (3, 0.03), arrowprops=props)
i = t_vals >= transformed
ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0')
i = t_vals <= -transformed
ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0')
ax.set_xlim(-5, 5)
ax.set_ylim(0, 0.1)
plt.show()
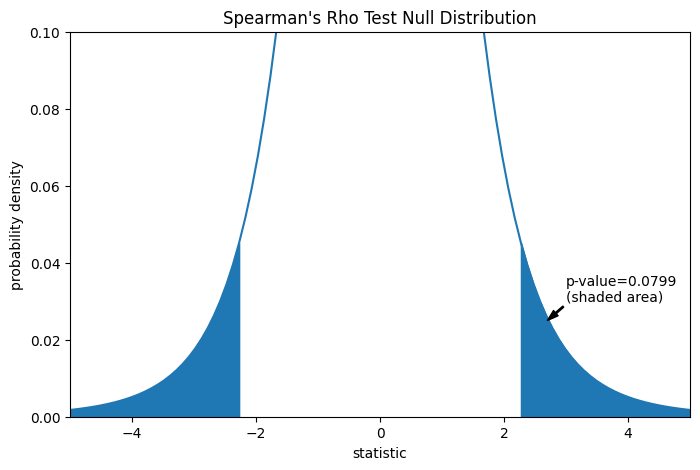
res.pvalue
np.float64(0.07991669030889909)
Wenn der p-Wert "klein" ist – das heißt, wenn die Wahrscheinlichkeit, Daten aus unabhängigen Verteilungen zu entnehmen, die einen so extremen Statistikwert ergeben, gering ist –, kann dies als Beweis gegen die Nullhypothese zugunsten der Alternative gewertet werden: Die Verteilungen von Gesamt kollagen und freiem Prolin sind *nicht* unabhängig. Beachten Sie, dass
Das Umgekehrte gilt nicht; d.h. der Test wird nicht verwendet, um Beweise für die Nullhypothese zu liefern.
Der Schwellenwert für Werte, die als "klein" betrachtet werden, ist eine Wahl, die getroffen werden sollte, bevor die Daten analysiert werden [3], wobei die Risiken von sowohl falsch positiven Ergebnissen (fälschlicherweise Ablehnen der Nullhypothese) als auch falsch negativen Ergebnissen (Nichtablehnen einer falschen Nullhypothese) berücksichtigt werden.
Kleine p-Werte sind kein Beweis für einen *großen* Effekt; sie können nur einen Beweis für einen „signifikanten“ Effekt liefern, was bedeutet, dass sie unter der Nullhypothese unwahrscheinlich aufgetreten wären.
Angenommen, die Autoren hatten vor der Durchführung des Experiments Grund zu der Annahme einer positiven Korrelation zwischen den Messungen von Gesamt kollagen und freiem Prolin und hatten sich entschieden, die Plausibilität der Nullhypothese gegenüber einer einseitigen Alternative zu bewerten: Freies Prolin hat eine positive ordinale Korrelation mit Gesamt kollagen. In diesem Fall werden nur die Werte in der Nullverteilung, die größer oder gleich der beobachteten Statistik sind, als extremer betrachtet.
res = stats.spearmanr(x, y, alternative='greater')
res.statistic
np.float64(0.7000000000000001)
fig, ax = plt.subplots(figsize=(8, 5))
plot(ax)
pvalue = dist.sf(transformed)
annotation = (f'p-value={pvalue:.6f}\n(shaded area)')
props = dict(facecolor='black', width=1, headwidth=5, headlength=8)
_ = ax.annotate(annotation, (3, 0.018), (3.5, 0.03), arrowprops=props)
i = t_vals >= transformed
ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0')
ax.set_xlim(1, 5)
ax.set_ylim(0, 0.1)
plt.show()
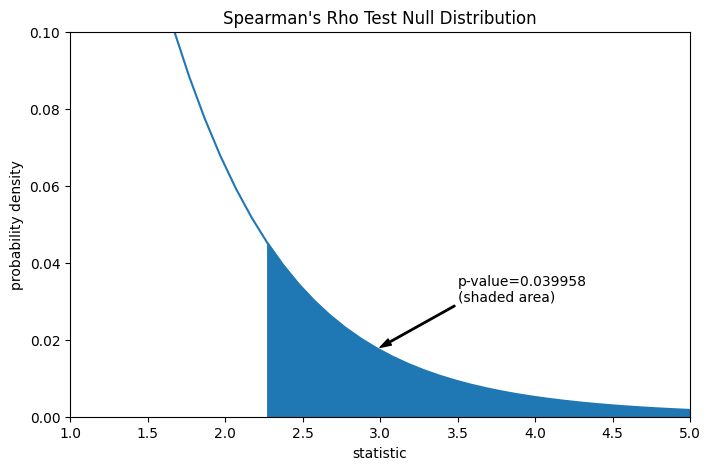
res.pvalue
np.float64(0.03995834515444954)
Beachten Sie, dass die t-Verteilung eine asymptotische Annäherung der Nullverteilung darstellt; sie ist nur für Stichproben mit vielen Beobachtungen genau. Für kleine Stichproben kann es angebrachter sein, einen Permutationstest durchzuführen [4]: Unter der Nullhypothese, dass Gesamt kollagen und freies Prolin unabhängig sind, war jede der freien Prolinmessungen gleich wahrscheinlich mit jeder der Gesamt kollagenmessungen beobachtet worden. Daher können wir eine *exakte* Nullverteilung bilden, indem wir die Statistik für jede mögliche Paarung von Elementen zwischen x und y berechnen.
def statistic(x): # explore all possible pairings by permuting `x`
rs = stats.spearmanr(x, y).statistic # ignore pvalue
transformed = rs * np.sqrt(dof / ((rs+1.0)*(1.0-rs)))
return transformed
ref = stats.permutation_test((x,), statistic, alternative='greater',
permutation_type='pairings')
fig, ax = plt.subplots(figsize=(8, 5))
plot(ax)
ax.hist(ref.null_distribution, np.linspace(-5, 5, 26),
density=True)
ax.legend(['asymptotic approximation\n(many observations)',
f'exact \n({len(ref.null_distribution)} permutations)'])
plt.show()
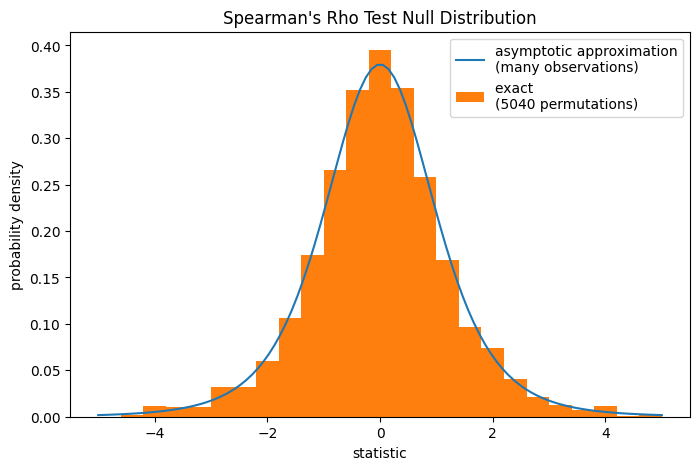
ref.pvalue
np.float64(0.04563492063492063)